Introduction
In this manuscript, we demonstrate the viability of a revised and heavily truncated PCR primer design for pan-microbial 16S rRNA gene amplification. This modification of common pan-microbial primers is based on three observations: (1) the high melting temperature of the widely used 515F primer, (2) reduced tolerance for mismatches at 3’ ends of primers,1–3 and (3) primer designs for 16S rRNA gene amplification often increase degeneracy but rarely alter the terminal 3’ position of the primer.4 We previously targeted the 515F and 806R primers for alteration,5 primarily by removing bases at the 5’ ends to introduce nucleotide diversity into amplicons for Illumina sequencing purposes. The very high melting temperature of the 515F primer suggested the possibility of more aggressively shortening the primer. Moreover, we hypothesized that shortening the primer would not reduce the target range of a pan-microbial primer, and could conceivably expand the target range, while still preserving specificity to the microbial 16S rRNA gene. By selective shortening of the primers, primers could be synthesized with differing 3’ termini. If the standard primers amplify some templates poorly due to mismatches at 3’ ends, then removing those 3’ ends might allow the newly designed primers to bypass such limitations. However, the truncated primers will introduce new 3’ mismatches with a different set of genomic DNA templates. To address this, we synthesized a series of staggered or cascading primers with varying 3’ ends to overcome constraints by any single 3’ end mismatch, without adding sequence degeneracies (Figure 1A). Novel primers and primer pools were compared to standard pan-microbial primers using PCR with complex templates (soil, feces, wastewater, and skin) followed by deep sequencing of amplicons.
Materials and Methods
Oligonucleotide primers
The Earth Microbiome Project (EMP) primers, the 515 forward primer (referred to as ‘515F’) and 806 reverse primer (referred to as ‘806R’), were used in PCR amplification of genomic DNA (gDNA) as the standard primer set.4,6–8 These primers target the V4 variable region of microbial 16S small subunit (SSU) ribosomal RNA (rRNA) genes. Melting temperatures were estimated using the OligoAnalyzer Tool 3.1 calculator,9 assuming primer concentrations at 250 nM, 2 mM Mg2+, and 0.2 mM dNTPs. When designing primer pools containing truncated primers (i.e., tr515F and tr806R), each unique primer sequence was added to the pool in equal molar concentrations (Figure 1A and Supplemental Material S1). All oligonucleotide primers, whether pooled or individual, were synthesized as LabReady primers and normalized to 100 µM. All primers contained 5’ linker sequences as part of a two-stage PCR protocol for generating amplicons for next-generation sequencing (NGS). Linker sequences were custom linkers compatible with xGen Amplicon UDI primers (e.g., IDT part# 10009846). The IDT linkers were CTACACGACGCTCTTCCGATCT (sIDTP5) used with forward primers and CAGACGTGTGCTCTTCCGATCT (sIDTP7) used with reverse primers.
Genomic DNA Sampling and Extraction
Four types of genomic DNA (gDNA) samples were used as templates for 16S rRNA gene amplification, including rat feces, human skin, wastewater, and bulk soil. The rat fecal sample was a pool of DNAs from a study of rats treated with dextran sodium sulfate (DSS) and included DNA from control and DSS-treated rats. Fecal DNA was extracted using the chemagic DNA Stool 200 Kit H96 implemented on a chemagic 360 instrument (Revvity Health Sciences, Waltham, MA, USA). Skin swabs, pre-moistened with sterile PBS, were self-collected from multiple participants and pooled together into a composite sample in DNA/RNAshield (Zymo Research) prior to nucleic acid extraction. Skin DNA was extracted using a Fecal Microbiome DNA Kit implemented on a Maxwell RSC48 device (Promega; Madison, WI, USA). The collection of skin samples from participants was reviewed and approved by the Institutional Review Board (IRB) at Rush University Medical Center (25012702-IRB01). No written consent (waiver of documentation of informed consent) was required as part of the consent process as the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context. Verbal consent from participants was received. Wastewater samples were collected from effluent of a local Long-Term Acute Care Hospital (LTACH) over the course of one month, and DNA was extracted using a QIAamp PowerFecal Pro DNA Kits and automated using a QIAcube device (Qiagen). A composite wastewater sample was created by pooling genomic DNAs from multiple samplings prior to PCR amplification. Soil samples were collected from various locations associated with wheat plants across Israel, and DNA was extracted using a DNeasy PowerSoil Pro kit (Qiagen; Hilden, Germany). A composite soil DNA sample was created by pooling genomic DNAs from multiple locations prior to PCR amplification.
Amplicon Library Preparation and Sequencing
For all experiments, a standard two-stage PCR amplification method was used to generate barcoded amplicons for sequencing.10 First stage PCR amplifications were performed with gene-specific primers (e.g., 515F/806R or tr515F/tr806R) containing 5’ IDT linker sequences in 10 µL reactions in 96-well plates, using repliQa HiFi ToughMix (QuantaBio, Beverly, MA, USA) as described previously.1,11 Genomic DNA inputs were standardized to one ng per reaction for all template types. Final primer pool concentrations were 400 µM. All reactions were performed with four to eight technical replicates, along with reagent blank negative controls. The first-stage PCR thermal cycling conditions were an initial denaturation at 98 °C for 30 seconds, followed by 28 cycles of denaturation at 98 °C for 10 seconds, annealing at 52 °C for 5 seconds, and elongation at 68 °C for 1 second.
Subsequently, a second-stage PCR, used for barcoding, was performed in 10 µL reactions in 96-well plates with repliQa HiFi ToughMix mastermix. Each well received a unique dual-indexed primer pair (xGen Amplicon UDI primers). Final concentrations of primers were 300 nM and each reaction used 1 µL of unpurified PCR product from the first stage as input. The second-stage PCR thermal cycling conditions were an initial denaturation at 98 °C for 30 seconds, followed by 8 cycles of denaturation at 98 °C for 10 seconds, annealing at 60 °C for 1 second, and elongation at 68 °C for 1 second.
After amplification, barcoded second-stage PCR amplicons were pooled together in equal volumes and purified using an AMPure XP cleanup protocol (0.7X, vol/vol; Beckmann Coulter, Inc; Brea, CA, USA). Pooled amplicons were purified twice using a 0.7X AMPure cleanup to ensure complete removal of fragments shorter than 300 base pairs. Cleaned amplicons were sequenced on an Illumina MiniSeq sequencer with a mid-output flow cell and employing 2 × 154 base reads with a 10% PhiX spike-in. Based on the results of the MiniSeq sequencing run, samples were re-pooled to balance output among samples.10 Pooled products were cleaned as described above and sequenced on an Illumina NovaSeq6000 flow cell with 2 x 259 base reads with a 15% PhiX spike-in. Library preparation and MiniSeq sequencing were performed at the Genomics and Microbiome Core Facility (GMCF) at Rush University (Chicago, IL). NovaSeq6000 sequencing was performed at the DNA Services Lab, Roy J. Carver Biotechnology Center, at the University of Illinois at Urbana-Champaign.
Bioinformatic Analysis of Sequence Data
Raw FASTQ files were merged using the software package PEAR12 using default parameters. Merged sequences were imported into QIIME213 and trimmed to remove adapters and those sequences shorter than 240 bases using cutadapt trim-single.14 The data was then denoised with dada2 denoise-single.15 Denoised sequences were filtered to exclude unwanted taxa, including Mitochondria, Chloroplast, Unassigned, and Eukaryota, using the taxa filter-table plugin. Finally, the remaining sequences were classified with the feature-classifier classify-sklearn plugin,16 utilizing a pre-trained SILVA classifier.17 Data sets with pooled standard primers and pooled truncated primers were rarefied in R18 to 50,000 and 300,000 reads for skin and soil composite DNA samples, respectively. Data sets with individual truncated forward primers and the standard 806R primer pool were rarefied to between 100,000 and 240,000 depending on sample type (skin, soil, feces, and wastewater). Features were filtered to a minimum sample type relative abundance of 1/10P5P before furth using prcomp. PERMANOVA analyses were performed using vegdist, and adonis2 from the vegan package.19 PERMDISP analyses were performed with Betadisper and permutest, also in the vegan package. Wilcoxon-Mann-Whitney, and Pearson correlations were performed using base R. Packages used for data organization and plotting included tidyverse,20 ggplot2,21 gridExtra,22 ggh4x,23 and ggtext.24
Data Sharing
Raw sequence data (FASTQ) files were submitted in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under BioProject PRJNA1198036.
Results and Discussion
Redesign of pan-bacterial 16S rRNA gene primers
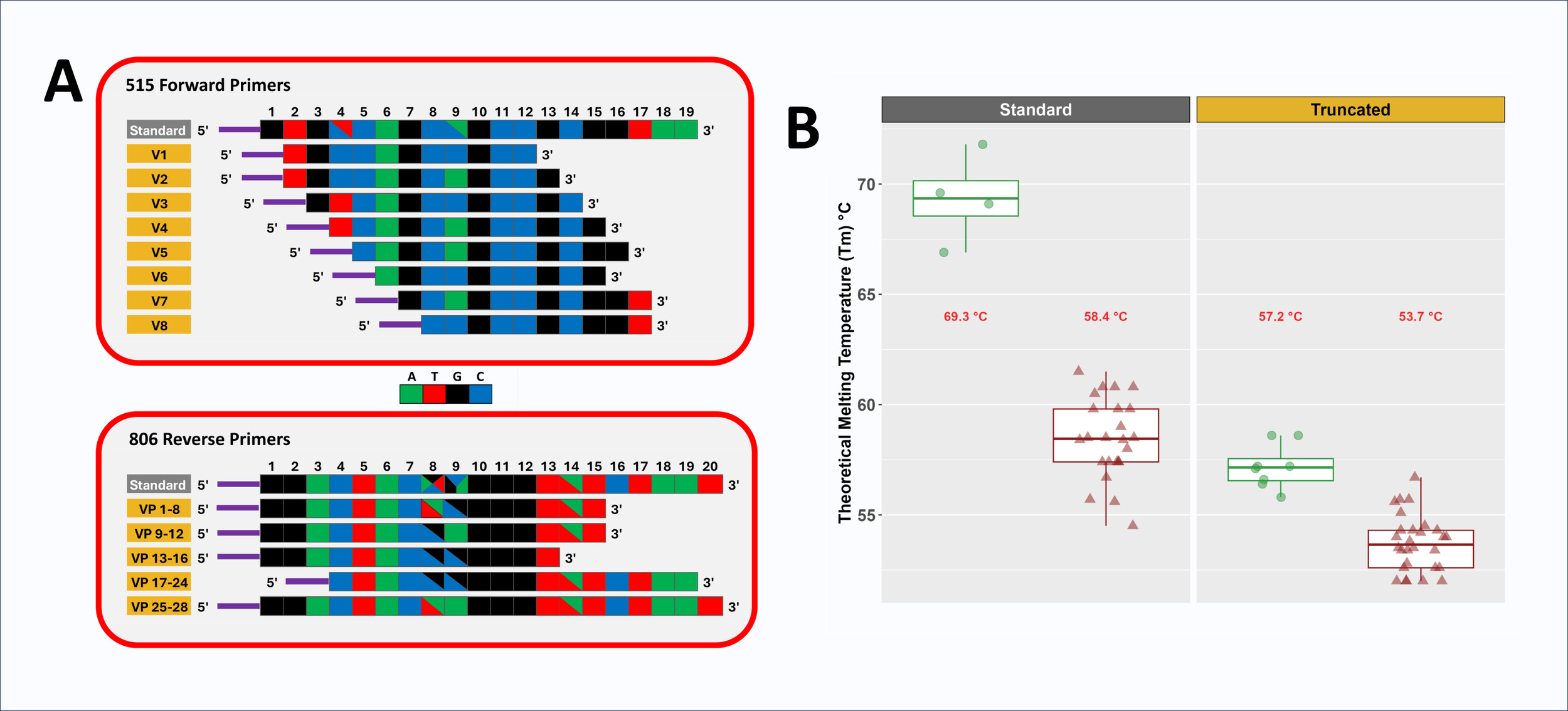
Domain-level primers targeting microbial 16S rRNA gene are typically degenerate to accommodate sequence variation across lineages, even in highly conserved regions. For example, the commonly used 515F primer 44 is four-fold degenerate with two degenerate positions, while the 806R primer is 24-fold degenerate with three degenerate positions (Figure 1A and Supplemental Material S1).4 We previously altered the 515F and 806R primers to reduce variation in melting temperature (Tm) among primer variants and to introduce nucleotide diversity into amplicons by removing bases from the 5’ ends.5 However, these prior modifications limited primer length reduction to 16 bases and focused on the 5’ ends, leaving a large disparity in average Tm between 515F and 806R primer pools.5 In the current study, we more aggressively trimmed bases from the original 515F primers to generate primers with melting temperatures more similar to that of the 806R primers. Due to the initial high Tm of the original 515F pool, primers as short as 10 bases were created in this study. Short primers have been developed previously,25 but in that study required custom polymerases to allow for low temperature annealing for PCR. The very high Tm of the original 515F primer allowed for length reduction while still retaining a melting temperature viable for standard PCR. In addition, the primer redesign in this study focused on 3’ end modifications to address inhibitory primer-template mismatches located at 3’ ends of primers.1 For example, the standard 515F/806R primer set poorly amplifies bacteria from the genus Cutibacterium (formerly Propionibacterium) due to single mismatches on each primer close to or at the 3’ end of primers.26,27 To address such issues, primer design aimed for a set of primers with a cascade of terminal 3’ positions (Figure 1A and Supplemental Material S1). Primer redesign proceeded with the following aims: (1) 515F primer Tm within 5 °C of the average Tm of the 806R primer pool, (2) primers positioned within the regions defined by the standard 515F and 806R primer regions (i.e., no bases beyond the 5’ or 3’ ends), (3) all degeneracies from the original primers maintained in the final primer pool, and (4) removal of degenerate positions from individual primers when possible. A key assumption of these modifications is that shortening the primers would not reduce the target range of the primer pair, as previously discussed.5,25
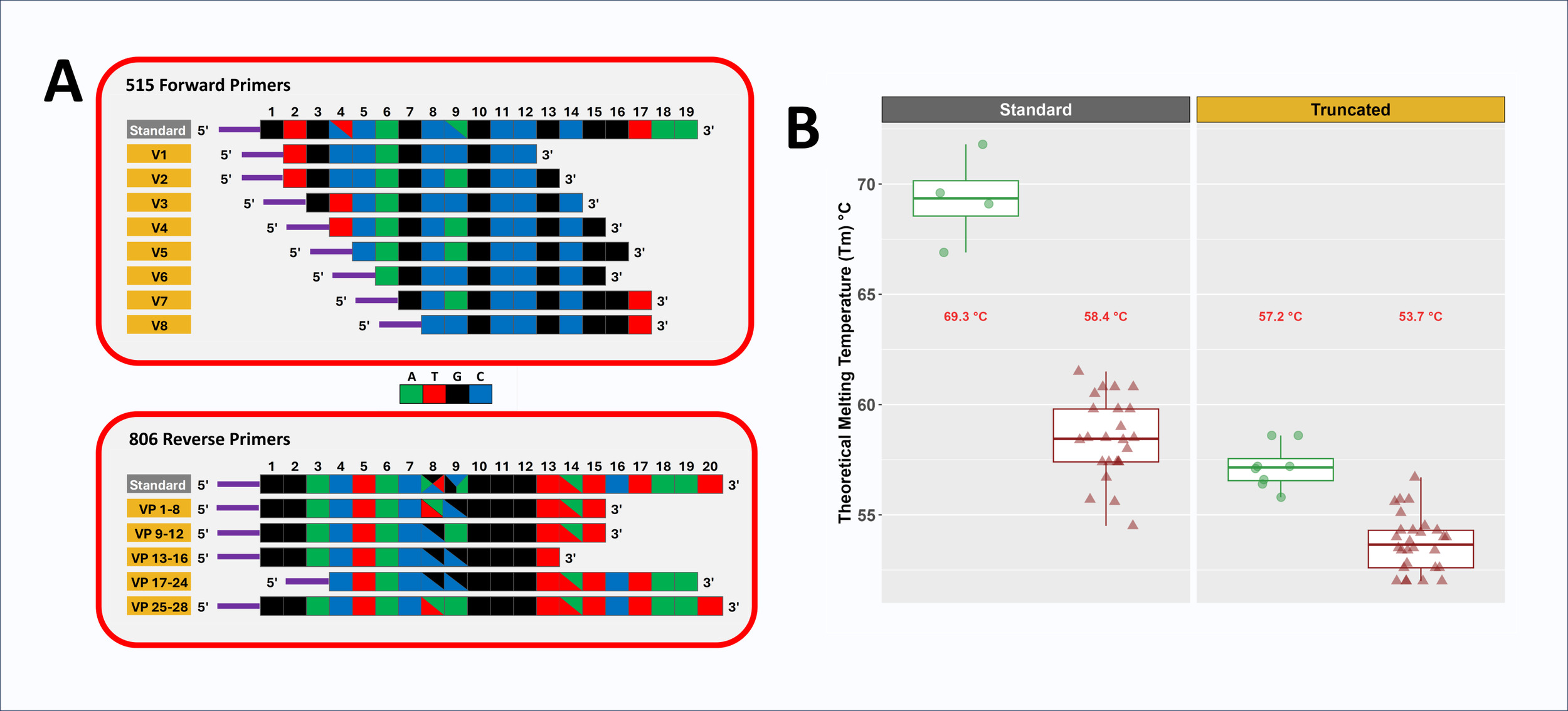
Two new pools of primers were developed for the 515F and 806R primer sets (Figure 1 and Supplemental Material S1). The original 515F primer pool (4-fold degenerate; Tm 66.9 to 71.8 °C; 19 bases) was altered by creating a cascade of eight non-degenerate primers with lengths of 10-12 bases and Tm values ranging from 55.8 to 58.6 °C. The final truncated 515F (tr515F) primer pool was composed of eight equimolar primers with seven different 5’ ends and six different 3’ ends. The original 806R primer pool, with relatively low melting temperatures (i.e., 54.5 to 61.5 °C), had less capacity for truncation. It also contained three degenerate positions with 24 total variants (20 bases). The standard 806R primer pool was replaced by a pool of 28 different variants, and in total had two different 5’ positions and four different 3’ positions (Supplemental Material S1). Truncated 806R primer lengths ranged from 13 to 20 bases, with Tm values ranging from 52 to 56.7 °C. Primers were pooled in equimolar ratio (i.e., each degenerate primer was pooled at a concentration commensurate with the level of degeneracy) to generate the final tr806R primer pool.
All primers were synthesized with 5’ linker sequences compatible with xGen Amplicon UDI primers (dual index; IDT). These linker sequences are essential for the two-stage library preparation protocol employed. During the first PCR stage, 515F/806R or tr515F/tr806R primers with 5’ linkers are used to amplify genomic DNA (gDNA). In the second PCR stage, indexing primers with Illumina adapters, sample-specific dual barcodes, and 3’ linkers were used to amplify templates created during the first stage and generate sequencer-ready libraries. Conceptually, the linkers do not interact with genomic DNA templates during the first stages of PCR due to the absence of matching sequences upstream of the gene-specific portion of the primer. However, in initial testing of truncated primers, it was observed that an 8-base truncated 515F primer with a linker was able to correctly amplify gDNA templates together with the standard 806R primer pool (Supplemental Material S2). The same 8-base truncated 515F primer, when synthesized without the 5’ linker, was unable to amplify genomic DNA templates, presumably due to the low melting temperature. Although the 8-base primer with linker was tolerated in a small test, all primers used in comparative analyses presented below were 10 bases or greater to ensure consistent on-target amplification and for purposes of pairing melting temperatures with the 806R primer pool. We infer that the linkers increase primer-template binding stability through sporadic base-pairing, upstream of the gene-specific portion of the primer.
Microbiome Profiling using 515F/806R and TR515F/TR806R primer sets
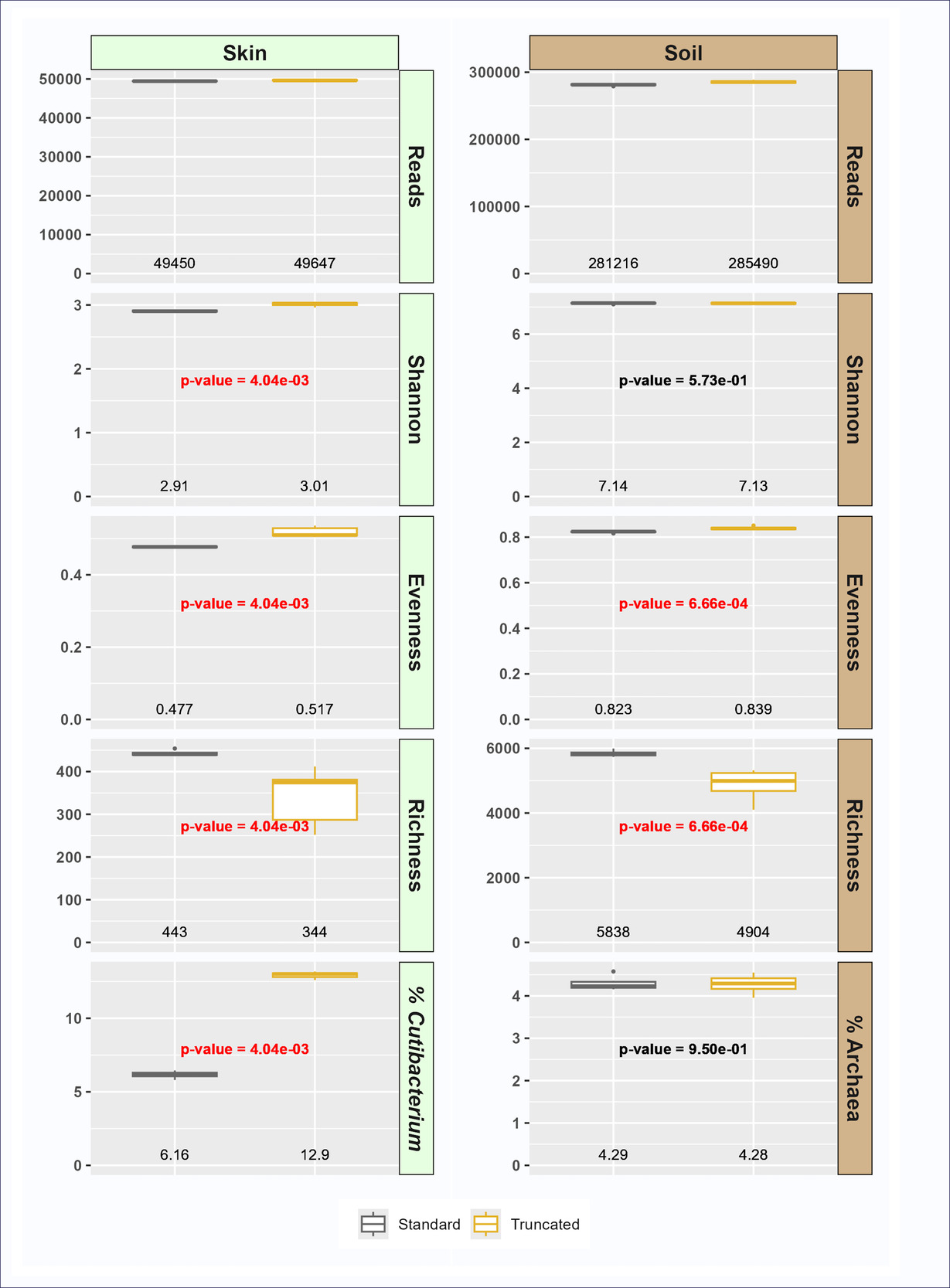
The objective of this study was to assess whether the use of truncated primer pools in PCR-NGS-based microbiome characterization alters the observed microbial community in complex samples. We hypothesized that truncated primers would generate microbial community profiles similar to or expanded from the standard primers, and that the truncated primer pools would enhance amplification of specific microbial taxa with known mismatches with the standard primers (e.g., Cutibacterium). Genomic DNA from skin and soil was amplified with the standard (515F/806R) and truncated (tr515F/tr806R) primer sets. These amplicons were deeply sequenced, and the resulting data were annotated and compared (Figures 2-4). After sequencing and annotation, datasets were rarefied to 50,000 sequences per skin sample and 300,000 sequences per soil sample. The datasets were then filtered by sample type for features with a minimum representation of 1/10P5P reads to remove variability due to very low abundance features or spurious features generated through PCR or sequencing error before analysis. All analyses were performed with 4-8 replicates.

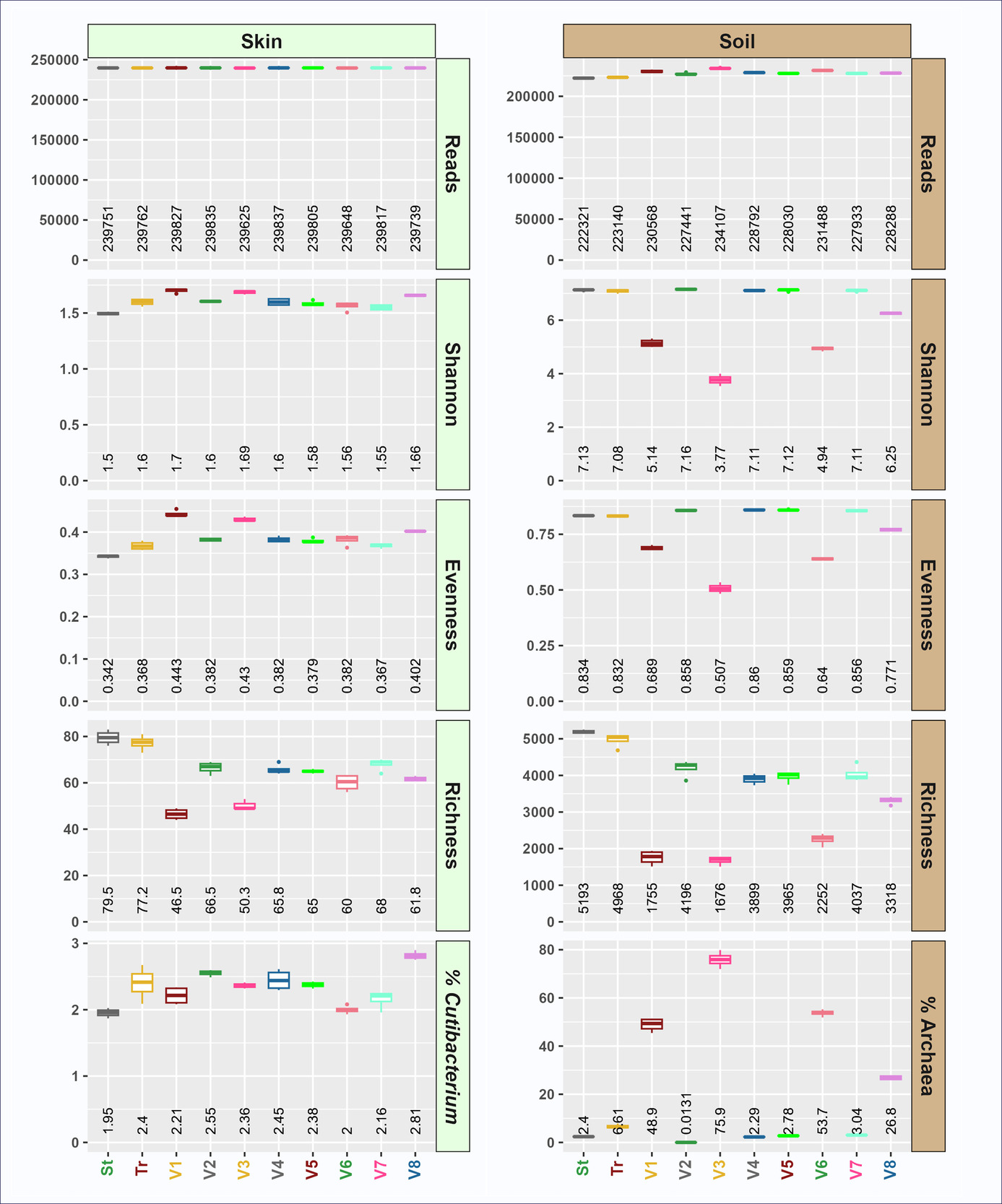
To evaluate the impact of the redesigned primers on sample characteristics, metrics for alpha diversity and representative taxa were calculated. Although overall alpha diversity was similar between primer sets, several trends were observed. First, taxonomic richness at the ASV level was significantly lower with the truncated primer set relative to the standard primer set (Figure 2). Conversely, evenness was significantly higher in the truncated primer set, resulting in Shannon indices that were not significantly different between primer sets (soil) or slightly higher in truncated primer set data (skin) (Figure 2). The relative abundance of sequences from the domain Archaea was not significantly different between primer sets in soil, but the relative abundance of Cutibacterium was significantly higher in the truncated primer set (12.9%) relative to the standard primer set (6.11%) in skin (Figure 2). Bacteria from the genus Cutibacterium (formerly Propionibacterium) have known mismatches with the standard 515F/806R standard primer, with mismatches either at the 3’ end (806R primer) or near the 3’ end (515F primer).28,29 The presence of mismatches between primer and template, especially mismatches close to the 3’ end of primers, has been shown to significantly reduce PCR amplification of target DNAs, leading to underestimation of the true abundance of the target.1,30 In this study we demonstrate that by truncating primers at 3’ ends and providing a cascade of primers with different 3’ ends, we can both overcome difficulty in detecting Cutibacterium while maintaining overall similarity of the total microbial community. The truncated primers are better able to amplify Cutibacterium because almost all primers in the tr515F/tr806R pool do not contain the 3’ bases with mismatches to the Cutibacterium template. This resulted in a doubling of the relative abundance of Cutibacterium in skin samples while still maintaining similar levels of overall diversity (e.g., Shannon index; Figure 2). The truncated primers do not appear to have negative impacts on systems without Cutibacterium, such as soil.

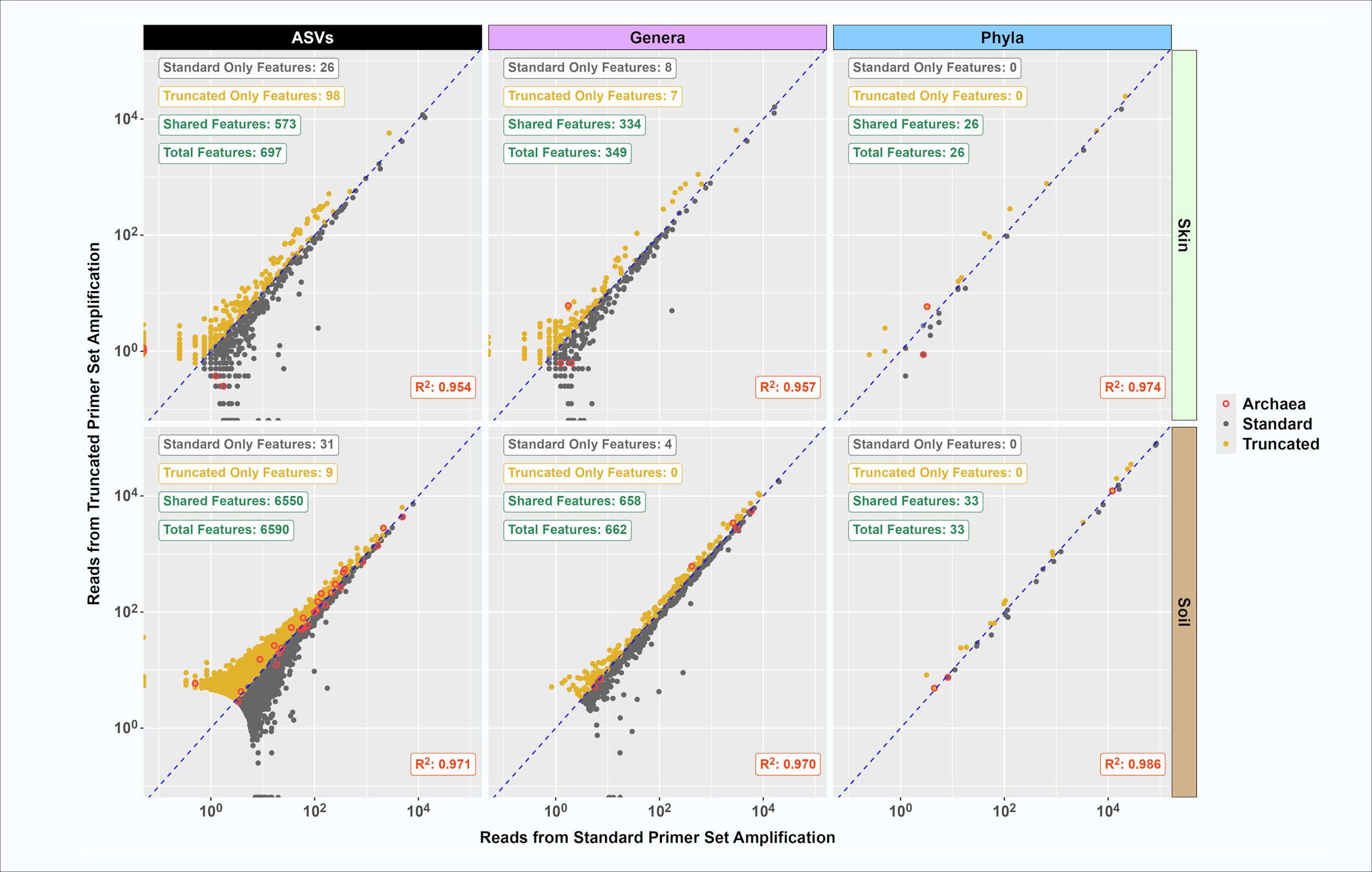
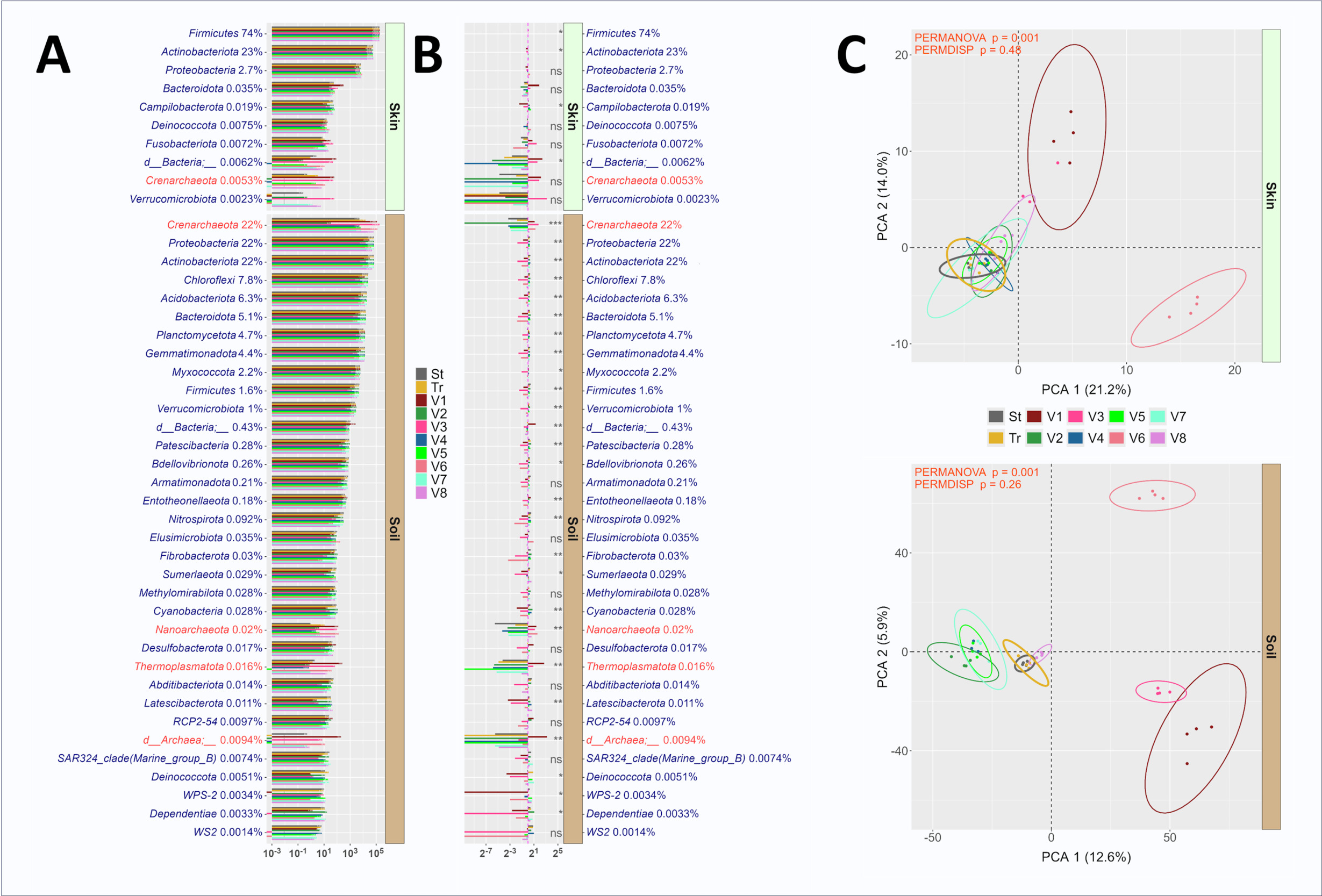
To look more closely at similarities in observed microbial communities characterized using standard and truncated primer sets, the average relative abundance of each taxonomic feature (ASV, genus and phylum) from the standard and truncated primer sets was plotted (Figure 3). High correlation values (RP2P>0.95) were calculated for skin and soil at ASV, genus, and phylum taxonomic levels. Most features were shared between the primer sets, with relatively few features found only with standard or only with truncated primer sets. For example, out of 6,590 ASVs observed in the composite soil sample, 31 were found only with the standard primer set while 9 were only found with the truncated primer set. In the composite skin sample, the truncated primer set detected more unique features relative to the standard primer set. Specifically, 98 unique ASVs were identified with the truncated primer set as compared to 26 unique ASVs with the standard primer set out of a total of 697 ASVs (Figure 3). There were fewer genus-level unique features detected (i.e., 8 skin genera uniquely detected with the standard primer set as compared to 7 unique genera detected with the truncated primer set and 4 unique soil genera detected with the standard primer set versus no unique genera detected with the truncated primer set). No unique phyla were detected between standard and truncated primer sets in either skin or soil (Figure 3).
_and_truncated_(tr515f_tr806r)_.png)
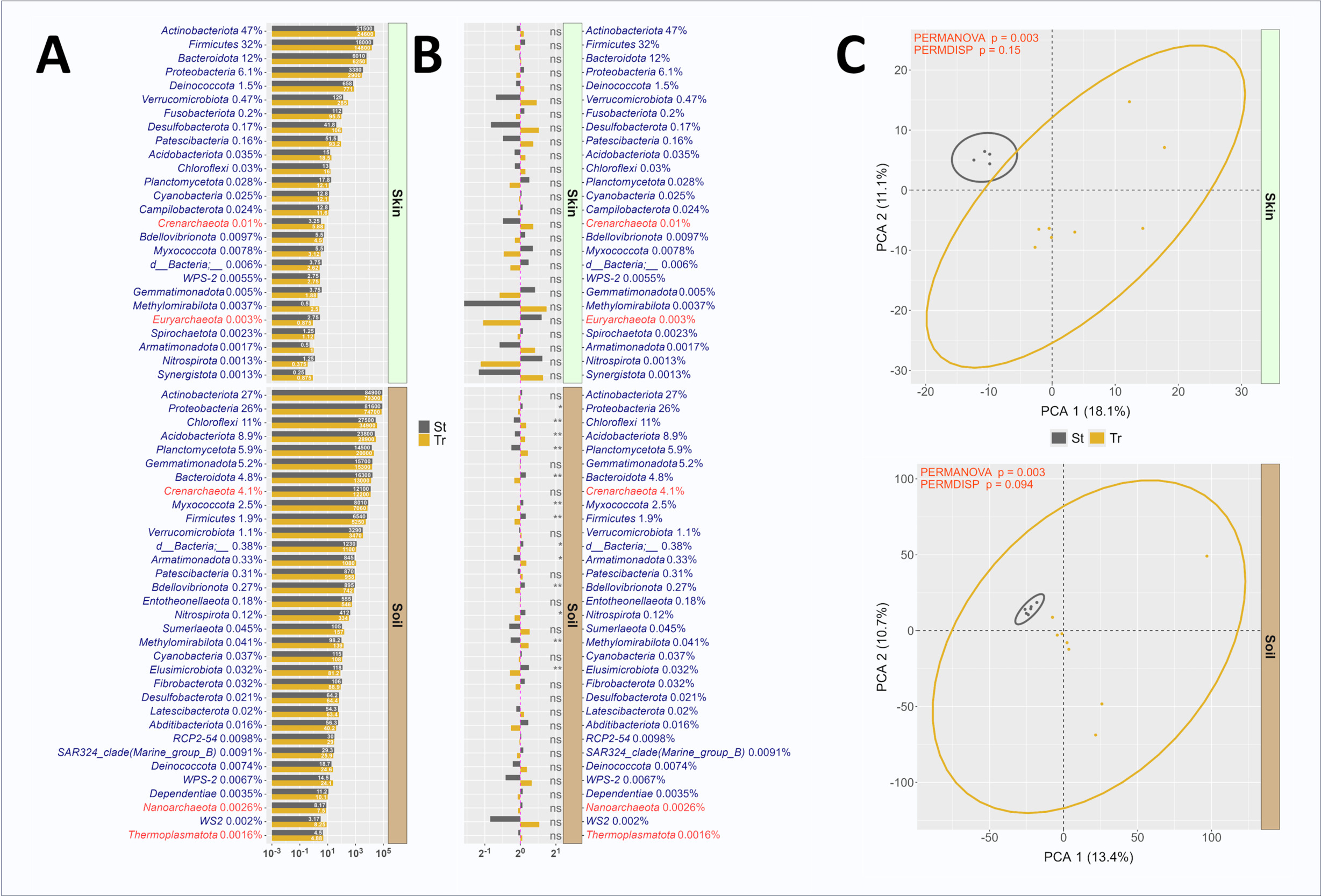
The differential abundance of microbial phyla was compared between standard and truncated primer sets (Figure 4A, 4B). In skin microbiomes, all phyla were present in sequence data generated with both primer sets, and no significant differences were observed in relative abundance between methods (Figure 4B). Similarly in soil, dominant phyla had similar relative abundance across the standard and truncated data sets, though the relative abundances of a number of phyla were significantly different, including Chloroflexi (higher in truncated), Acidobacteriota (higher in truncated), Planctomycetota (higher in truncated), Armatimonadota (higher in standard), and Methylomirabilota (higher in truncated). PERMANOVA analysis demonstrated small but significant differences in overall microbial community structure as observed with standard and truncated primer sets. Differences in dispersion between standard and truncated linker primer set profiles was trending towards significance in both skin and soil microbiomes, as measured through PERMDISP (Figure 4C), and this reflects the greater observed variability in replicates for the truncated primer set relative to the standard primer set. Although some differences between primer sets were observed, these results indicate a marginal effect of the truncated primer set on observed microbial communities. Impressively, the 515F/806R primer sites appear to be highly tolerant of manipulation without substantive modification to the amplified templates. This was observed both in this study and our prior efforts to introduce nucleotide diversity for Illumina sequencing largely through removal of bases at the 5’ ends of primers.5
Microbiome Profiling using non-degenerate truncated 515F primers with standard 806R primers
The role of each truncated 515F primer variant (V1 through V8; Figure 1A) was explored by performing PCR amplifications together with the standard 806R primer pool as the reverse primer (Figures 5 and 6; Supplemental Materials S3 and S4). All sample types were analyzed with 3-4 replicates and rarefied to a depth of 100,000 to 240,000 sequences per sample, depending on sample type. Samples were then filtered for features with a minimum representation of 1/10P5P reads.
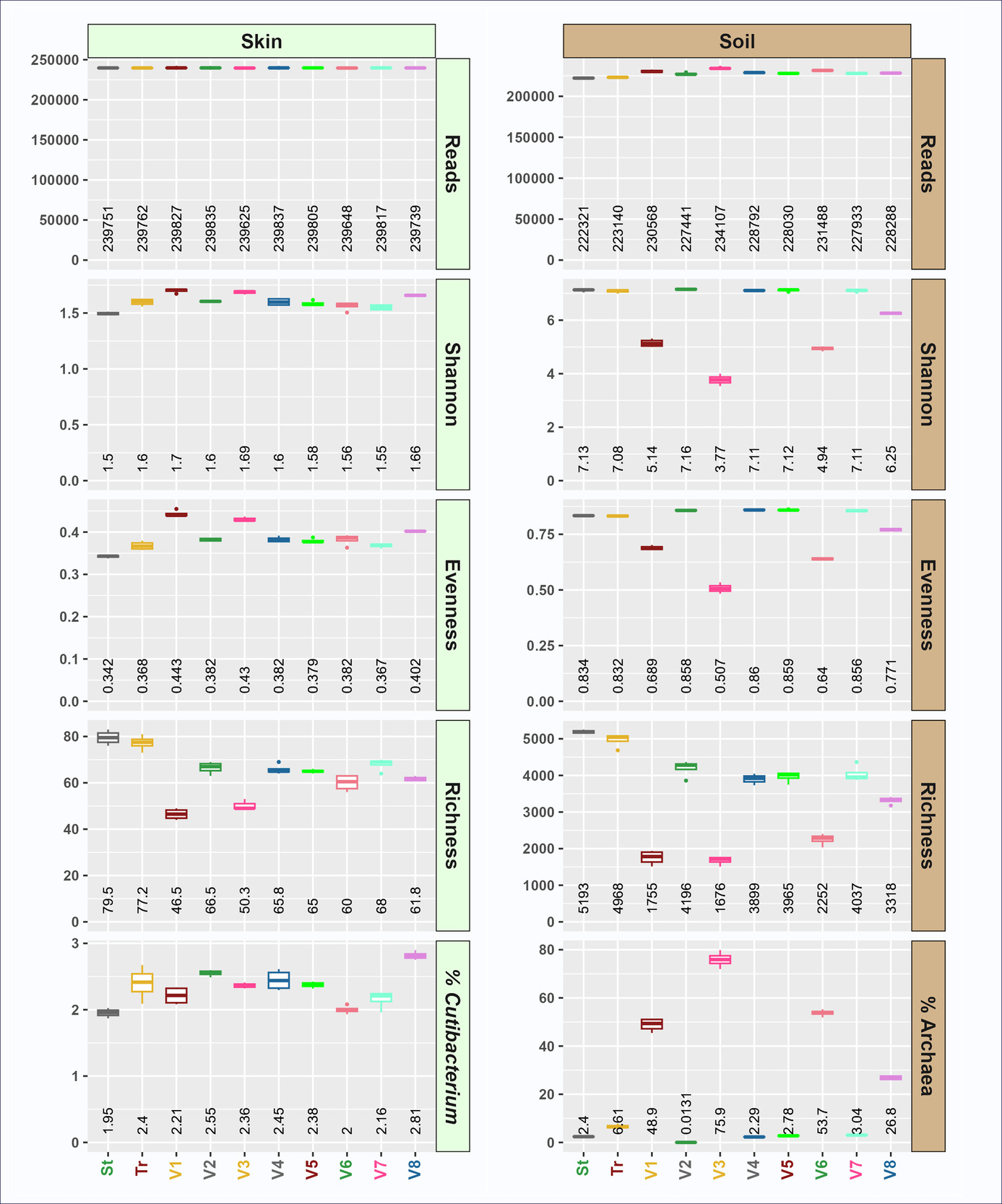
Using non-degenerate truncated forward primers yielded marked differences in alpha (Figure 5; Supplemental Materials S3) and beta (Figure 6; Supplemental Material S4) diversity. These primers yielded divergent alpha diversity indices, with reduced taxonomic richness in soil (Figure 5). Although soil microbial evenness was slightly increased using four of the tr515F variants (variants 2, 4, 5, and 7), microbial evenness was dramatically decreased in the remaining tr515F variants (variants 1, 3, 6, and 8). Soil Shannon indices tracked with richness and evenness, with variants 2, 4, 5, and 7 having Shannon indices similar to the standard pool and truncated pool of primers, while variants 1, 3, 6, and 8 had severely reduced Shannon indices. Furthermore, we observed that the relative abundance of Archaea was dramatically higher in soil reactions with primer variants 1, 3, 6, and 8 (ranging from 26.8 to 75.9%) as compared to 0.0131 to 3.04% in reactions with primer variants 2, 4, 5, and 7 (Figure 5). Average archaeal relative abundance in the same soil sample ranged from 2.4% (standard pooled primers) to 6.61% (truncated pooled primers) when pooled 515F or tr515F primer sets were used (Figure 5).
The relative abundance of archaeal sequences in the skin gDNA sample was too low for robust analysis. However, a similar phenomenon was observed in the relative abundance of Archaea for wastewater (Supplemental Material S3), with truncated primer variants 1, 3, 6 and 8 again substantially enriched for Archaea. Briefly, the relative abundance of Archaea in wastewater using standard or truncated forward primer pools ranged from 0.0023 to 0.016%, while the relative abundance of Archaea in reactions with forward primer variants 1, 3, 6, and 8 ranged from 0.107 to 0.598% (Supplemental Material S3).
In contrast to the marginal effects of the pooled primers, the individual truncated primer variants exhibit distinct characterizations. Notably variants 1, 3, 6, and 8 yielded dramatically elevated relative abundance of sequences annotated as Archaea (Crenarchaeota, Nanoarchaeota and Thermoplasmatota) when amplifying soil gDNA (Figure 6A, 6B) consistent with trends observed in Figure 5. Of this Archaeal-enriching group, variants 1, 3, 6 (shades of red) strongly diverged from pooled primers (St and Tr) and other individual variants (Figure 6C). The remaining variants (2,4,5,7) either converged with the pools (Skin) or diverged slightly (Soil).
__truncated_(tr515f_806r)__and_.png)
All four of the primers enriching for archaeal sequences contained a “C” at the second degenerate position in the standard 515F primer variants 1, 3, 6, and 8 (i.e., Position 9, Figure 1A and Supplemental Material S1; position underlined: GTGYCAGCMGCCGCGGTAA). Among the four archaeal-enriching truncated primer variants, primer variant 3 (GTCAGCCGCCGC) yielded the highest relative abundance of Archaea in soil with over 75% of sequences annotated as Archaea. Previous studies have developed archaeal-specific primers targeting the same region as the 515F primer. For example, an early study highlighted primer PARCH519r/probe (S-D-Arch-0519-a-S-15; shown in the inverse complement to the 515F primer - TTACCGCGGCKGCTG) that spans the same region as primer variant 3.31,32 The degenerate position “K” (i.e., G or T) in the PARCH519r corresponds to the same position indicated by the degenerate “M” (i.e., C or A) in the forward orientation of the primer. Our findings demonstrate that the replacement of the degenerate base with a single “C” in the 515F primer significantly alters the relative abundance of archaeal sequences when archaeal DNA is present in the genomic DNA pool. This is consistent with another earlier primer design S-D-Arch-0516-a-S-25, which also lacked degenerate base at the indicated position and instead used the same “C” identified in this study, TGYCAGCCGCCGCGGTAAHACCVGC),32 and was used as an oligonucleotide probe in archaeal studies.33,34 Interestingly, the other degenerate position in the 515F primer (i.e., Position 4, Figure 1A and Supplemental Material S1; position underlined: GTGYCAGCMGCCGCGGTAA) was developed in part to increase Archaea coverage,35,36 and the truncated primer variant that contains the archaeal-enriching base “T” at this position (truncated variant 3) has the highest relative abundance of Archaea of any of the truncated variants. Overall, the identification of an archaeal-specific variant of the commonly used 515F primer may be beneficial for the microbial ecology community and is unique for the elevated relative abundance of archaeal sequence data despite standard PCR conditions and the high abundance of bacterial DNA in the sample. While the standard 515F/806R primer set can detect both bacteria and Archaea in general microbial community analyses (e.g., Earth Microbiome Project), focused primer sets are more frequently applied for archaeal analyses. For example, the V3-V4 region has been previously targeted with primers ARC344F/ARC806R37, and the V6-V8 region has been targeted with primers ARCH915F/ARCH1386R38. Finally, in analyses of soil with individual forward primer variants, the strong enrichment in Archaea with primers containing the “C” base resulted in lower microbial alpha diversity (richness, evenness, and Shannon Index). However, when these archaeal-enriching primers were pooled with non-enriching primers (e.g., truncated 515F pool), no significant increase in the relative abundance of Archaea across the entire dataset was observed (Figure 2). Thus, the truncated variants 1, 3, 6 and 8 of the 515F primer set likely need to be separated from the other primers to produce an increase in the relative abundance of Archaea. Pooling the truncated primer variants 1, 3, 6 and 8 together could provide a cascading primer pool of 515F primers that will heavily enrich for Archaea in combination with the standard or truncated 806R primer pools.
Conclusion
The design and redesign of pan-microbial 16S rRNA gene primer sets have been common activities for microbial ecologists for decades. Primer optimization involves balancing the use of highly conserved regions of the gene for primer design while ensuring primers are sufficiently long for robust and gene-stringent amplification. Longer primers provide for greater gene-specific stringency but decrease the potential target range by increasing the likelihood of mismatches with templates in metagenomic DNA from complex microbial communities. Mismatches can be overcome by incorporating degeneracy into primer design, but excessive degeneracy is not always well tolerated for broad-range and accurate PCR amplification. We sought to increase the target range of pan-domain-level primers by shortening primers and creating staggered, truncated primers with varying 3’ ends. Due to the sensitivity of PCR to 3’ mismatches,1 the staggered approach was intended to mitigate 3’ mismatches affecting any specific taxonomic groups. Our findings indicate that the staggered, truncated primer pools were viable for microbiome characterization generating microbial profiles highly similar to those observed with the standard, non-truncated primer pools. In skin microbiome, the truncated primer pools increased the observed relative abundance of Cutibacterium due to the removal of known 3’ mismatches between Cutibacterium and the standard 515F/806R primer set. More broadly, however, the truncated primer pools did not markedly increase the target range in the samples tested and produced results equivalent to the standard primer pools with differences largely manifesting in very low abundance features. However, when individual truncated primer variants were tested, a single nucleotide position in the 515F primer site was observed to yield sequence datasets that were dramatically enriched in archaeal sequences. The results of this study demonstrate there is greater design flexibility in primer design than has previously been considered. This versatility may be highly advantageous in the development of primers for microbial functional genes which tend to be long and have high degeneracy due to third-position codon degeneracy.39–41 The tolerance of PCR for short primers (tested as short as 8 bases in this study) is in part a result of the very high melting temperature of the standard 515F primer, but also due to the 5’ linkers used in two-stage PCR protocols, as employed in this study. These linkers, despite lacking sequence similarity to DNA templates, appear to interact with the DNA templates in a limited fashion leading to increased priming complex stability and in turn to elevated melting temperatures. Our findings highlight the value of primer redesign as shown by improved detection of Cutibacterium in skin microbiome samples and demonstrate the viability of short primers for amplicon library preparation for NGS.
Acknowledgements
We gratefully acknowledge the support of the members of the Genomics and Microbiome Core Facility (GMCF; Rush University, IL, USA). Soil samples were collected as part of a US-Israel Binational Agricultural Research and Development Fund grant (IS-5537-22; Stefan Green, co-PI). Rat feces were collected as part of NIH-funded research (R21AR075130-02; Rick Sumner, PI). Wastewater was collected as part of a Centers for Disease Control and Prevention (CDC) / SHEPheRD Contract 75D30121D12772 (Michael Lin, PI).
Statement of financial support/COI
None
Human subjects statement
The collection of skin samples from participants was reviewed and approved by the Institutional Review Board (IRB) at Rush University Medical Center (25012702-IRB01). Verbal consent from participants was received.